渐渗片段思路整理
一、核心算法设计
-
滑动窗口机制
- 固定窗口大小:
- 窗口滑动规则:
- 起始条件:从染色体第一个SNP开始,当前窗口首SNP必须匹配参考基因组(基因型为'0/0')
- 终止条件:窗口最后一个位点必须匹配参考基因组(基因型为0/0)
- 匹配比例:窗口内匹配参考基因组(达尔文棉)的SNP数量 > 5 × 匹配目标样本(泗棉3号)的数量
- 窗口移动策略:
- 满足条件时:跳跃式移动(跳过当前窗口,直接检查后续30个SNP)
- 不满足条件时:步进式移动(每次移动1个SNP位置)
-
sliding_window()- 核心匹配逻辑:
for 窗口 in 染色体SNP序列: 统计 matches_darwin = 当前样本'0/0'计数 统计 matches_target = 与目标样本相同基因型计数 if matches_darwin > 5*matches_target and 窗口末SNP=='0/0': 记录候选区域
- 核心匹配逻辑:
-
处理流程:
graph TD A[开始窗口位置i] --> B{检查位置i的基因型} B -->|0/0| C[分析整个30 SNP窗口] B -->|非0/0| D[滑动到i+1] C --> E[统计匹配参考基因组数量] C --> F[统计匹配目标样本数量] E --> G{满足条件1?} F --> G G -->|是| H{窗口末端基因型=0/0?} G -->|否| D H -->|是| I[记录区域并跳至i+30] H -->|否| D
二、v1.1 区域合并算法
- 合并条件:
- 位于同一染色体
- 相邻区域间距 ≤ 合并阈值(默认5M)
- 合并操作:
- 更新区域结束位置为后一区域的结束位置
- 累加匹配参考基因组数量
- 累加匹配目标样本数量
- 过滤条件:合并后区域长度 ≥ 最小长度阈值(默认500K)
- 处理流程:
graph LR A[排序区域列表] --> B[初始化合并列表] B --> C[遍历所有区域] C --> D{与上一区域同染色体且间距≤阈值?} D -->|是| E[合并区域] D -->|否| F[添加新区块] E --> C F --> C C --> G[完成遍历] G --> H[过滤长度不足区域]
二、v1.2区域合并算法
-
双层合并机制
graph LR A[原始窗口] --> B[窗口合并] --> C[片段合并] --> D[最终结果]
- 窗口合并阶段:
- 合并相邻窗口(默认间距≤500K)
- 累加匹配计数
- 片段合并阶段:
- 合并相邻片段(默认间距≤1M)
- 过滤短片段(v1.2.2-500k是去掉小于500K、v1.2.2-5M是去掉小于5M)
- 窗口合并阶段:
-
merge_regions()- 合并策略:
按染色体位置排序区域 遍历区域列表: 若当前区域与上一区域间距≤合并阈值: 扩展上一区域的end位置 累加matches计数 否则: 创建新区域 过滤长度<min_length的区域
- 合并策略:
三、结果输出
- 排除目标样本(泗棉3号)
- 对每个样本独立分析
- 双文件输出:
_tmp.xls:原始窗口结果(未合并区域)- Chromosome Start End:染色体、起始位置、结束位置
- Matches:匹配达尔文氏棉的snp数量(样品和达尔文氏棉相同)
- Simian3 Matches:匹配泗棉三号的snp数量(样品和泗棉三号相同)
_results.xls:最终合并结果- Chromosome Start End:染色体、起始、终止位置
- Length (Mb):区域长度(Mb)
- SNP Count:匹配达尔文氏棉的snp数总和
- SNP per Mb:匹配达尔文氏棉的SNP密度(每Mb数量)
- SNP Count(Simian3):匹配泗棉3号的SNP总数
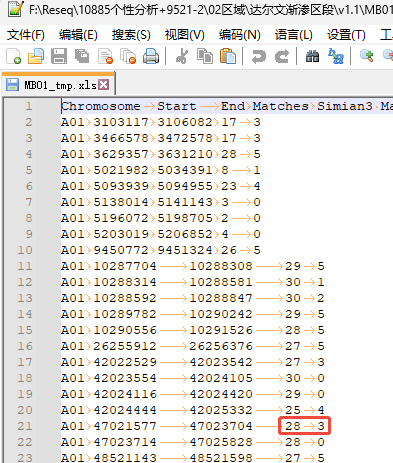
文章里“并获得每个窗口双亲同源 SNP 数目的比值,即达尔文氏棉亲本同源 SNP 数目:泗棉 3 号亲本同源 SNP 数目,将比值大于 25:5 的窗口定义为渐渗窗口”,
也就是30个snp窗口内,(与达尔文氏棉相同的位点)大于5倍的(与泗棉三号相同的位点),上图(28)大于(5倍的3)。